








Dr Kapur is an Associate Professor and Executive Director of Cardiovascular Center for Research and Innovation Tufts Medical Center in Boston. His research focuses on acute and chronic heart failure, circulatory support device development, and cardioprotective mechanisms in the setting of acute myocardial infarction. Dr Kapur is founding member of the A-CURE Working Group, and Co-Chair of the A-CURE Symposium.
Dr Kapur began by presenting an overview of the history of left ventricle (LV) unloading over the past decade. He noted that mechanical devices developed in recent years have provided hope for heart failure (HF) patients who previously had no options. The mechanical devices that have been employed for unloading have developed from biventricular devices (BVADs) and HeartWare® HVAD® pumps, where the aim was cardiac replacement as bridge to transplant or destination therapy, to an increasing use of percutaneous technologies such as the Impella pumps, where the goal is cardiac recovery and not replacement. He highlighted that percutaneous heart pumps have given clinicians the chance to promote the recovery of a patient’s native heart.
He then presented a brief history of the A-CURE movement, which began in Boston in 2015 and has since hosted meetings in Paris and Rome prior to this meeting in Barcelona. In those 2 years, research has progressed rapidly from preclinical testing to clinical trial launch. However, despite considerable progress in the field of LV unloading, questions remain, notably whether we can reduce the burden of ischaemic HF after a myocardial infarction (MI), and what are the cardioprotective mechanisms underlying LV recovery.
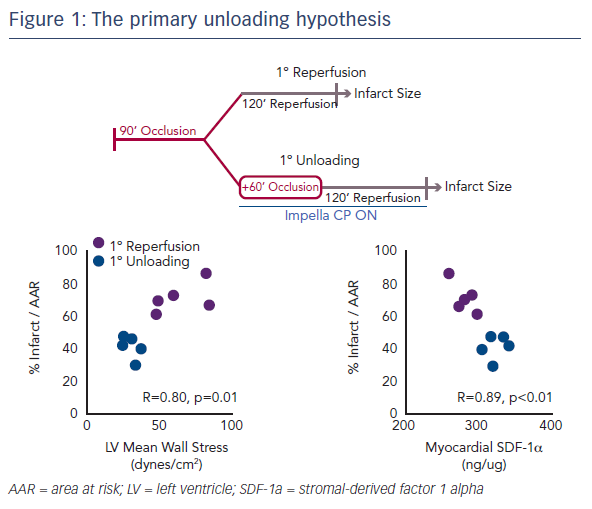
Myocardial infarct size remains an important target of therapy.1 However, even if infarct size is reduced following an MI, if the haemodynamics are consistent with HF, this will remain a major cause of mortality for patients.2 Two years ago, Dr Kapur’s team published the concept of the primary unloading hypothesis, which suggested that first unloading the LV, then delaying reperfusion, activates a cardioprotective programme that limits myocardial damage in acute MI (Figure 1).3 This study also identified an early molecular signal, release of the cytokine stromal-derived factor 1 alpha (SDF-1-alpha), which is known to be cardioprotective. This correlated with infarct size and led to the hypothesis that mechanical unloading leads to an increase in the SDF-1 CXCR4 signalling pathway, which is linked to a number of other cardioprotective mediators, including protein kinase B, extracellular signal-regulated kinase and glycogen synthase kinase 3 beta. This results in a shift to a cardioprotective phenotype.
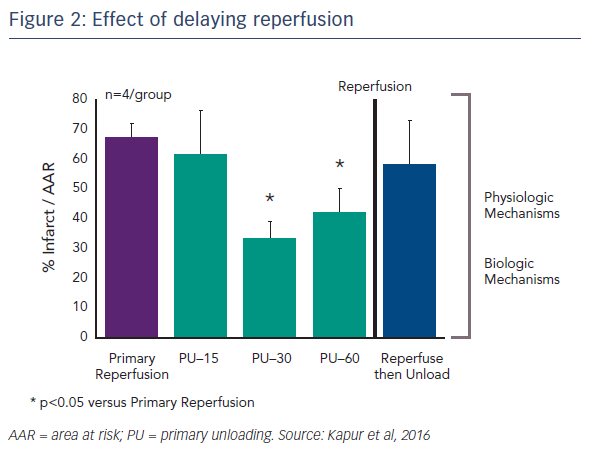
Another important question concerns the kinetics of primary unloading: how important is the delay to reperfusion? Dr Kapur’s team tested the idea of delaying reperfusion after activating the Impella device by 15, 30 or 60 minutes. Delaying reperfusion appeared to be necessary for reducing infarct size (see Figure 2).4 One possible reason for this is that functional reperfusion may reduce the area of risk. With the left anterior descending artery (LAD) still occluded, enhanced collateral flow through non-occluded vessels may lead to a reduction in the area at risk. This may, in part, drive the benefits in terms of reducing infarct size.
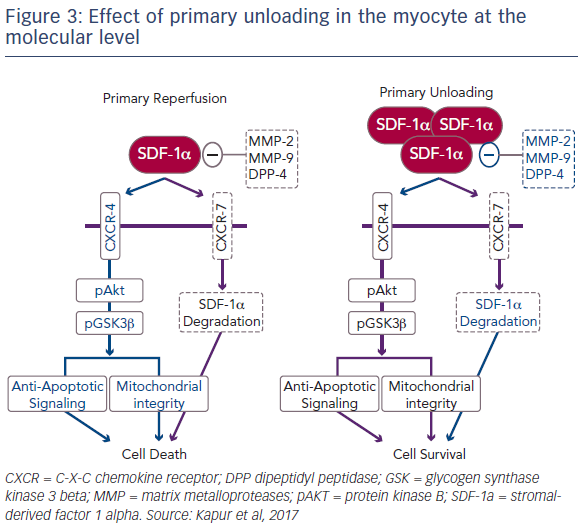
The release of SDF-1-alpha in ventricular tissue is highest after a 30-minute delay in reperfusion. In order to fully understand the biological mechanisms underlying unloading, it was important to explore the cause of this release. SDF-1-alpha is ubiquitously expressed, but is rapidly degraded by a number of metalloproteases as well as dipeptidyl peptidase-4 (DPP4) and the CXCR7 pathway.5 Further study revealed that primary unloading reduces the activity of these proteases that promote SDF-1-alpha degradation. Dr Kapur’s team is currently investigating the hypothesis that, by reducing the activity of these degradation pathways, primary unloading can increase the concentration of SDF-1- alpha in the myocardium, particularly during acute injury, leading to a protective phenotype that increases cell survival (Figure 3).6 It is known that ischaemic injury leads to an uncoupling of SDF-1-alpha and CXCR signalling.7 Dr Kapur suggested that primary unloading re-aligns the SDF- 1-alpha:CXCR signalling axis, which is vital for myocardial repair.
An important question to address was whether the acute cardioprotective effect of primary unloading provides a durable reduction in HF. The cardiac response to increased work includes a reactivation of foetal genes, and remodelling following acute MI is largely driven by the foetal gene programme.8 An animal study found that, compared to primary reperfusion, primary unloading reduces LV scar and preserves cardiac output at 30 days after acute MI.6 No early signs of change in cardiac volume were seen but this may be due to the short timescale of the study. The study also showed that, at 30 days, primary unloading limits the activation of a gene programme associated with maladaptive cardiac remodelling. It also reduces tissue expression and circulating levels of brain natriuretic peptide, an important marker of HF, and increases the circulating levels of SDF-1-alpha in the first week, which correlates directly with reduction of scar size. Primary unloading appears to mechanically reprogramme myocardial responses to injury in acute MI, which involves the foetal gene programme.9
In summary, research to date has provided a platform for further investigation. Administration of primary unloading and stabilising haemodynamics following acute MI offer the potential for interventions that have until now been considered impossible. These include the administration of adjunct pharmacotherapy during an anterior STEMI including intravenous beta blockade, intracoronary vasodilators, glucose, insulin, potassium, SDF-1-alpha, protease inhibitors, and neuromodulation. Current research is investigating the administration of intravenous esmolol during primary unloading to increase the oxygen supply:demand ratio.
Dr Kapur concluded by reminding the audience that, in 2015, it was predicted that mechanical preconditioning would not translate into a successful clinical strategy that reduces myocardial infarct size.10 In 2017, the US Food and Drug Administration approved a Phase I clinical trial examining the safety and feasibility of primary unloading.