








Dr. del Rio is a Research Scientist at MyoKardia in San Francisco, California. He was the recipient of the Best in Research Scholarship for the 2017 A-CURE Symposium.
Dr Del Rio presented data from his investigation into how mechanical support may fundamentally alter the mechano-energetic relationship in the heart. He began by providing the background to his study. By design, durable intra-cardiac left ventricular assist devices (LVAD) support the systemic circulation and cardiac output by removing blood from the left ventricle (LV), resulting in preserved systemic pressures and decreased stroke work (SW), stroke volume, filling pressures and preload. Unfortunately, these beneficial effects have not translated to recovery of heart function in these patients. In questioning this, Dr Del Rio examined the determinants of oxygen consumption in the left heart, particularly contractility and haemodynamic load. Historically, researchers have assumed that the heart does not respond to its altered physiological state resulting from implantation of an LVAD. Dr Del Rio’s team proposed the hypothesis that LVAD support can lead to paradoxical increases in the effective arterial elastance (Ea) and intrinsic cardiac contractility during ventriculoarterial coupling, therefore hindering mechano-energetic unloading.1 This imposes an intrinsic barrier to achieving LVAD-mediated recovery/reverse remodelling.
Dr Del Rio described an experiment in which an LVAD was inserted into a healthy animal and provided chronic partial support (>70 % of cardiac output). Over the course of 7 weeks, rather than maintain a steady state of lower device-dependent LV end diastolic volumes, the preload increased despite LVAD support. The Ea also showed an acute increase that normalised over time as the LV end diastolic pressure increased. There was a concomitant increase in early contractility, increased ventricle fibrosis and early release of atrial natriuretic peptide (ANP). There was an acute increase in contractility and an increase in fibrosis of the ventricle. This suggested that chronic partial support in healthy animals may trigger LV remodelling.
A subsequent study assessed the acute effects of LVAD support on systemic haemodynamics, LV mechano-energetics, and myocardial oxygen consumption (MVO2) in vivo.2 The study involved 12 mixedbreed sheep (34 to 54 kg), which were given acute LVAD support. The study assessed MVO2, using coronary sinus/arterial sampling catheters and left circumflex artery [LCX] coronary flow probe), systemic/LV haemodynamics, cardiac output (pulmonary artery flow) and load-independent LV inotropy/lusitropy via pressure-volume relationships. A continuous-flow LVAD (RotaFlow) device was used. Energetic components were determined before and during LVAD unloading, at both partial (50 % support, aortic valve opening) and complete (100 % uncoupling) support). These were compared with data obtained during partial inferior vena cava occlusion (IVCX, n=8) at matched level of volume unloading. Data were also collected when phenylephrine was administered to restore systemic haemodynamics (IVCX+PE) in order to mimic partial support.
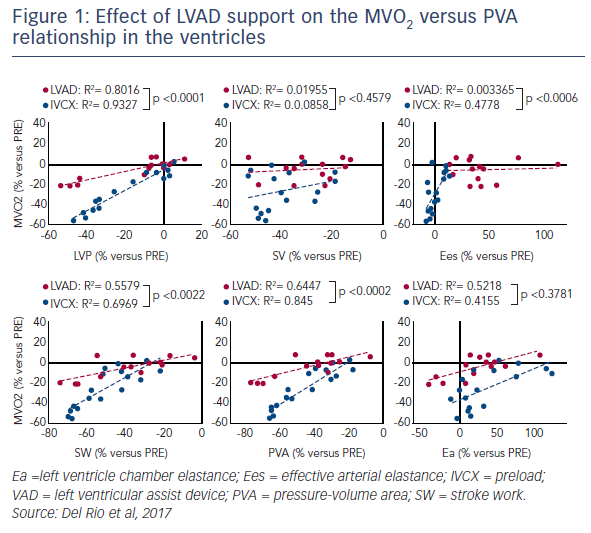
Results showed that partial LVAD support (57±4 % total cardiac output) preserved systemic/peak LV pressures (–3 ± 2 %) and cardiac output (–1±1 %), while decreasing LV preload (–13 ± 2 %), filling pressures (–29±7 %) and stroke volume (–28±5 %). Both the estimated LV chamber elastance (Ea; +40±11 %) and effective arterial elastance (Ees; +33±7 %) increased with support. The release of ANP was also reported during partial support. Despite marked reductions in SW (–29±5 %) and PVA (–31±4 %), there was a negligible change in MVO2 (+1±2 %). By contrast, complete support (109±9 %) decreased LV pressures (–33±10 %), normalised ANP release, and normalised Ea (–1±14 %) but not Ees (+54±12 %). There were further reductions in SW and PVA, with moderate MVO2 reductions (–13±4 %). Unsupported reductions in the preload (IVCX) decreased pressures. There was a decrease in MVO2 (–39±4 %), and PVA (–58±4 %). The Ea and Ees remained unchanged. Pressure support (CTRL+PE) increased Ea and blunted the MVO2 reductions (–7±2 %). Of interest, the LVAD support altered the MVO2 versus PVA relationship in the ventricles (see Figure 1).2
In conclusion, acute intra-cardiac LVAD support, particularly under partial unloading, can trigger mechano-energetic alterations, paradoxically hindering the ability of an LVAD to energetically unload the ventricle. There may be a limit to MVO2 reductions under LVAD support. Use of the LVAD engages intrinsic coupling mechanisms of the ventricles. Finally, an LVAD is, perhaps, perceived as a 'stress' signal, reflected in the release of ANP. On the basis of this research, Dr Del Rio said that there was a need for an increased understanding of the coupling and the mechanism that allows the heart to perceive the LVAD signal. This may allow us to pharmacologically inhibit this mechanism and increase the effectiveness of LVAD-mediated unloading on heart recovery. This should give us the beneficial effects of circulatory support as well as the potential for PVA reduction without a shift in the PVA/MVO2 relationship.