








Acute coronary syndromes (ACS) are a major cause of morbidity and mortality. Despite the use of optimal medical therapy and revascularisation there remains a significant risk of vascular events. Registry data indicates a persistent risk even in patients who are event free in the first year following ACS, with as many as 1 in 5 patients suffering a vascular event in the subsequent 3 years.1
The central process underlying ACS is the development of a thrombus overlying a ruptured or eroded plaque, leading to various degrees of acute vessel occlusion and myocardial ischaemia.2 A thrombus that originates following plaque rupture consists largely of platelets; in addition, coagulation pathways are also triggered by plaque rupture and platelet aggregation.3
Therapies that modify thrombogenesis form the foundation for the management of ACS and prevention of recurrent ischaemic events. The net clinical benefit of antithrombotic therapies must be weighed against the inevitable increased risk of bleeding.
This article will review the pathophysiology of thrombosis and evidence for the use of anticoagulants in ACS, including recommendations from the current European Society of Cardiology (ESC) guidelines.4
Pathophysiology of Thrombogenesis
Vascular damage triggers a cascade of pathways designed to maintain the integrity of the coronary circulation and to achieve haemostasis. Under normal conditions, controlled regulation of these pathways achieves the right balance between adequate coronary flow and appropriate vessel repair. Disruption of this homeostasis in the coronary circulation may result in life-threatening thrombosis.
Acute coronary syndromes are characterised by vascular inflammation, subsequent endothelial dysfunction and platelet activation, followed by thrombus formation.5 In the most extreme circumstances, uncontrolled thrombosis can culminate in complete vascular occlusion and ST-segment elevation MI (STEMI).6
Early mechanical and chemical reperfusion with percutaneous coronary intervention (PCI) and the use of antithrombotic agents respectively form the basis of ACS treatment and have been proven to reduce the frequency of both early and late cardiovascular events.7–13 Increased use of PCI further necessitates adequate antithrombotic therapy to reduce the risk of device-related complications.
Individual patient assessment is required to balance the need for thrombosis inhibition against a subsequently increased bleeding risk, which itself is an independent adverse prognostic marker in post-PCI patients.10,14
Mechanisms of Thrombus Formation
Activation of coagulation pathways is crucial for thrombus formation. Fibroblasts and smooth muscle cells express the membrane protein tissue factor, which is also present in blood. At sites of vascular damage, platelets express disulphide isomerase, which cleaves tissue factor into its active form. Activated tissue factor can then bind factor VIIa and the resulting complex activates factors VII, IX and X. Factors Xa and V complex together promoting thrombin generation.
The presence of thrombin activates factors V and VII promoting prothrombin conversion to thrombin by the more active complex Xa–Va. Fibrin generation from fibrinogen is triggered early in the coagulation cascade resulting in thrombus formation (Figure 1).15,16
Anticoagulation Therapies
The combination of anticoagulation with antiplatelet agents is more effective in reducing recurrent thrombotic events in non-ST elevation ACS (NSTE-ACS) than use of antiplatelets alone. This is due to the inhibition of thrombin production and activity.17
Unfractionated Heparin
Unfractionated heparin (UFH) is a sulphate-polysaccharide that is endogenously secreted. Its pentasaccharide component has a high affinity for antithrombin III (AT). Binding causes unfolding of antithrombin III exposing its active site more efficiently. The result is a dramatic increase in AT ability to inactivate thrombin and factor Xa.18
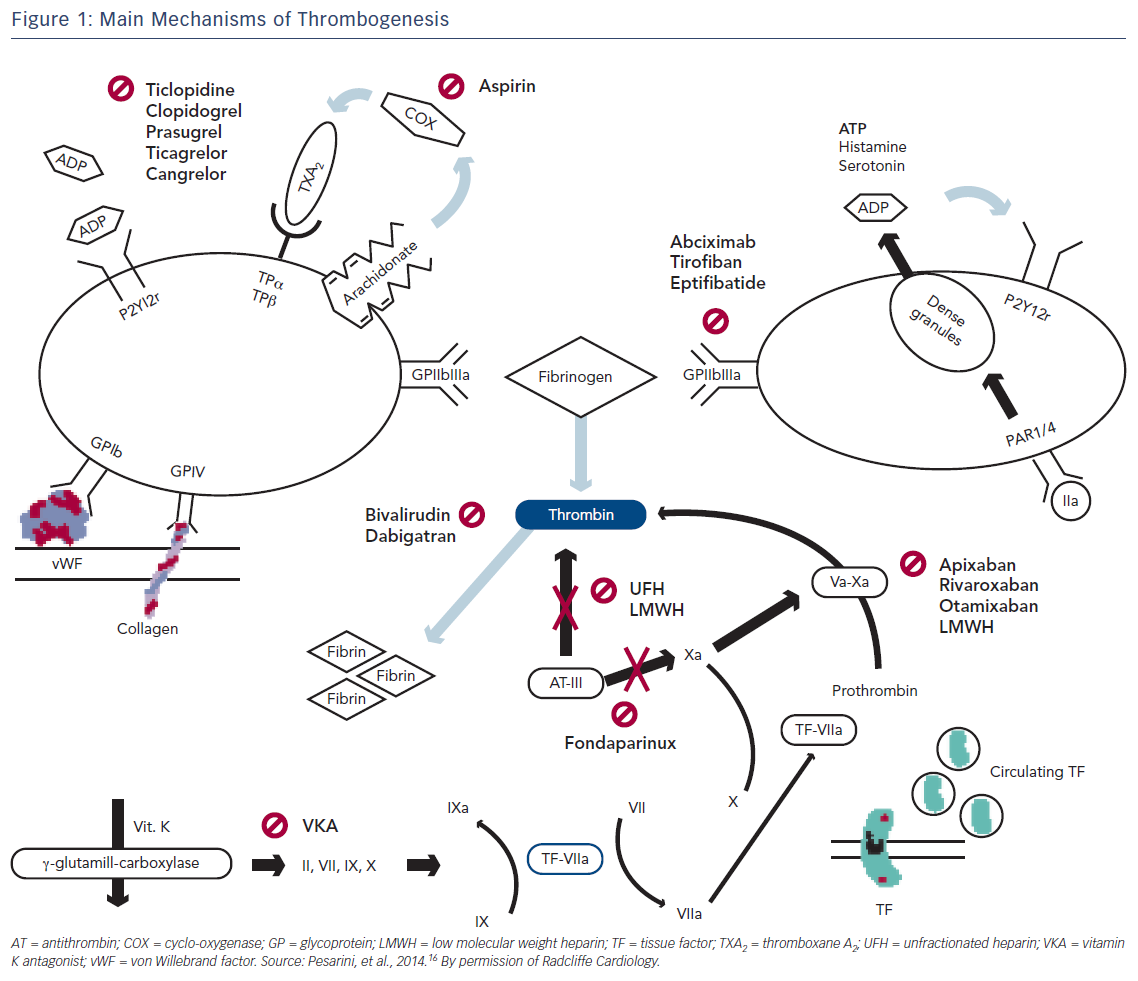
The narrow therapeutic window of UFH and its significant pharmacokinetic variability between patients requires administration to be closely monitored. Its anticoagulant effect can be monitored using either the activated clotting time (ACT) in the cardiac catheterisation laboratory or the activated partial thromboplastin time (aPTT) in other areas.
The efficacy of UFH in ACS has been validated in various randomised controlled trials.8,19–21 In summary, all trials consistently revealed a significant reduction in the frequency of recurrent ischaemic events. The Fondaparinux with Unfractionated Heparin During Revascularization in Acute Coronary Syndromes (FUTURA/OASIS-8) trial compared a low dose of UFH (50 IU/kg) against standard dosing (85 IU/kg) in patients with NSTE-ACS, and showed that dose adjustment had no significant effect on rates of major peri-PCI bleeding or vascular access-site complications.22
Intravenous UFH dosing is weight dependent, with current ESC guidelines recommending an initial bolus of 60–70 IU/kg up to a maximum of 5000 IU, followed by an infusion of 12–15 IU/kg/h up to a maximum of 1000 IU/h.4 During PCI, ACT-guided IV UFH boluses can be used, aiming for a range of 200–250 seconds if a glycoprotein IIb/IIIa (GPIIb/IIIa) inhibitor is given and 250–350 seconds in all other cases. Alternatively, weight-adjusted UFH at 50–70 IU/kg in combination with a GPIIb/IIIa inhibitor or 70–100 IU/kg (in the absence of GPIIb/IIIa) can be administered.4 If there are no other indications for UFH, it should be stopped following revascularisation.
The ESC recommends use of additional parenteral anticoagulation both before and after fibrinolysis in ST-elevation ACS (STE-ACS) with anticoagulation, and this should be used until planned definitive revascularisation is performed.23 Medically managed patients should be anticoagulated for at least 48 hours.
The use of UFH in patients with primary PCI (PPCI) has not been evaluated in placebo-controlled trials. It is, however, routinely recommended in patients not receiving bivalirudin or enoxaparin. An initial bolus of 70–100 U/kg is recommended when no GP IIb/IIIa inhibitor is planned. A dose of 50–60 U/kg should be administered when the use of GP IIb/IIIa inhibitors is expected.23 There is no clear evidence supporting ACT monitoring of UFH in the context of PPCI and doing so should not delay revascularisation.
The use of UFH poses a greater bleeding risk when compared with other anticoagulation strategies. Despite this, it remains popular, in part due to its efficacy in combination with low cost, short half-life and easy reversibility with protamine.24
Low Molecular Weight Heparin
Low molecular weight heparins (LMWHs) are 2–10 Kda derivatives of heparin that are well absorbed subcutaneously and have a longer half-life compared with UFH. They are less likely to bind to plasma proteins, thereby making the pharmacokinetics of LMWH more predictable than that of UFH, and reducing the likelihood of side effects such as bleeding and heparin-induced thrombocytopaenia (HIT).25–27
Enoxaparin is the most studied and utilised LMWH. Non-inferiority compared with UFH in patients with NSTE-ACS managed with aspirin and tirofiban was demonstrated in the A to Z trial.28 Enoxaparin was found to be non-inferior with respect to a composite end-point of death and non-fatal MI at 30 days in patients presenting with high-risk NSTE-ACS managed with an early invasive strategy in the Superior Yield of the New Strategy of Enoxaparin, Revascularization and Glycoprotein IIb/IIIa Inhibitors (SYNERGY) trial.29 A significant increase in the rate of TIMI major bleeding was noted in the enoxaparin arm compared with the UFH arm. However, in the Acute Myocardial Infarction Treated with Primary Angioplasty and Intravenous Enoxaparin or Unfractionated Heparin to Lower Ischemic and Bleeding Events at Short- and Long-term Follow-up (ATOLL) trial, rates of death, recurrent ACS and urgent revascularisation were significantly reduced in patients treated with enoxaparin (30 % versus 52 %; p=0.015), with no significant increase in bleeding rates.30
Enoxaparin is marginally favoured in a meta-analysis of all trials comparing the combined endpoint of death and MI at 30 days in patients with ACS receiving either enoxaparin or UFH (10 % versus 11 %; OR 0.90; 95 % CI [0.810–0.996]; p=0.043).31 At 7 days, no significant between-group difference in major bleeding rates was noted (6.3 % with enoxaparin versus 5.4 % with UFH; OR 1.13; 95 % CI [0.84–1.54]). Another meta-analysis of 23 trials involving 30,966 patients suggested superiority of enoxaparin in reduction in the rates of a composite of death and MI, complications of MI and bleeding when compared with UFH.24
In patients presenting with STE-ACS, the Assessment of the Safety and Efficacy of a New Thrombolytic 3 (ASSENT 3) trial compared outcomes of 6095 patients thrombolysed with tenecteplase receiving empirical enoxaparin versus UFH.32 Despite increased bleeding rates, the net clinical benefit favoured enoxaparin as rates of in-hospital recurrent ischaemic events were significantly lower in patients receiving enoxaparin up to a maximum of 7 days. Pre-hospital use of the same dose of enoxaparin in the ASSENT-3 PLUS trial was associated with a significant increase in rates of intracranial bleeding in elderly patients.33
In the Enoxaparin and Thrombolysis Reperfusion for Acute Myocardial Infarction Treatment-Thrombolysis in Myocardial Infarction Study 25 (ExTRACT–TIMI 25) trial lower doses of enoxaparin (0.75 mg/kg twice daily) in patients aged >75 years and those with significant renal impairment demonstrated lower rates of MI and death at 30 days compared with UFH (intravenous bolus of 60 U/kg of body weight followed by an infusion of 12 U/kg/h). Although rates of non-intracranial bleeding were significantly increased with enoxaparin, the net benefit favoured enoxaparin.34,35
Several non-randomised studies have also shown a clear benefit of enoxaparin over UFH in PPCI.24,36,37 In the ATOLL trial, enoxaparin (0.5 mg/kg IV followed by SC treatment) was compared with UFH.30 There was no significant reduction in the primary composite endpoint of death, MI, procedural failure and major bleeding at 30 days. However, reductions were noted in secondary composite endpoint of death, recurrent MI or urgent revascularisation, and in other secondary composite endpoints such as death, or resuscitated cardiac arrest and death, or complication of myocardial infarction were seen. Unlike previous studies, enoxaparin use was not associated with increased bleeding risk compared with UFH use in the PPCI setting.30
Subcutaneous enoxaparin at a dose of 1 mg/kg twice daily is the most frequently used anticoagulant in NSTE-ACS, as recommended by the ESC if fondaparinux is not available.4 It is contraindicated in patients with a glomerular filtration rate (GFR) <15 ml/min/1.73 m2, but the dose can be reduced to 1 mg/kg once daily for patients with a GFR of 15–29 ml/min/1.73 m2. In the latter case, it is advisable to monitor anti-Xa activity, which should also be done in patients whose body weight exceeds 100 kg. If the last enoxaparin dose was given ≥8 hours prior to PCI, a further 0.3 mg/kg IV bolus should be administered at the time of PCI.38,39 It is not advisable to change anticoagulant at the time of PCI.40
The ESC recommends that anticoagulation with enoxaparin may be used in preference over UFH peri-procedurally in patients with STE-ACS due to undergo PPCI.23,41–43
Fondaparinux
Fondaparinux is a selective Xa inhibitor with a half-life of 17 hours administered subcutaneously and once daily in patients with NST-ACS. It prevents the formation of thrombin by reversibly binding to antithrombin. Similarly to enoxaparin, fondaparinux rarely binds plasma proteins resulting in a more predictable anticoagulant effect, and no monitoring is required as it is fully bioavailable. Although there is no risk of HIT, fondaparinux is renally excreted and is not recommended if estimated GFR is <20 ml/min/1.73 m2.
In a dosing study, patients with ACS who were randomised to enoxaparin or varying doses of fondaparinux showed no relation of clinical endpoints with different fondaparinux dosing regimens leading to the establishment of the lowest dose – 2.5 mg.44
In the Arixtra Study in Percutaneous Coronary Intervention: a Randomized Evaluation (ASPIRE) trial, 350 patients undergoing PCI were randomised to receive either fondaparinux (2.5 mg or 5 mg) or UFH.45 There was no significant difference in rates of bleeding between the two groups (6.4 % versus 7.7 %; p=0.61), but significantly fewer bleeding events were noted when the lower dose (2.5 mg) of fondaparinux was used.
An analysis of 20,078 patients demonstrated non-inferiority of fondaparinux compared with enoxaparin with respect to ischaemic events in NSTE-ACS in the fifth Organization to Assess Strategies in Acute Ischaemic Syndromes (OASIS-5) study.46 The use of fondaparinux in this trial resulted in a substantial reduction in 30-day and 6-month mortality rates. In-hospital major bleeding rate was approximately half of that of the enoxaparin arm. The rate of major bleeding events at 9 days in patients who had PCI was significantly lower in those treated with fondaparinux compared with enoxaparin.47. This was independent of the timing of the intervention in relation to the last dose of anticoagulation administered. Catheter-related thrombosis occurred more frequently in patients pre-treated with fondaparinux leading to a recommendation to give a bolus of UFH at the time of PCI.
The findings from the OASIS trial were replicated in a real-world Scandinavian registry analysing 40,616 patients and showing reduced rates of bleeding and in-hospital death in patients treated with fondaparinux for NSTE-ACS when compared with LMWH.48
The use of fondaparinux in the context of primary PCI was associated with potential harm in the OASIS 6 trial and is therefore not recommended.23,49 In this trial, STEMI patients receiving streptokinase, rates of recurrent MI and death were significantly reduced in patients receiving fondaparinux compared with those treated with UFH or placebo.49,50
Due to its efficacy and safety profile, the ESC recommends the use of subcutaneous fondaparinux at a dose of 2.5 mg once daily in patients presenting with NSTE-ACS regardless of the planned management strategy unless coronary angiography is imminent.4,48 In patients managed for NSTE-ACS with fondaparinux a bolus of UFH is recommended at the time of PCI to reduce the risk of catheter-related thrombosis.22,51
Bivalirudin
Bivalirudin is a synthetic congener of naturally occurring hirudine, with a high affinity for thrombin in its clot-adherent and circulating form, thereby preventing the conversion of fibrinogen to fibrin. The bivalirudin–thrombin bonds can be gradually cleaved by thrombin itself making bivalirudin’s actions reversible. It has a short half-life of 25 minutes. Bivalirudin’s anticoagulant effect is predictable as it does not bind plasma proteins and monitoring can be done using APTT or ACT measurements. No association between bivalirudin and HIT has been found.
Outcomes for bivalirudin (0.75 mg/kg followed by 1.75 mg/kg/h during the intervention) plus GPIIb/IIIa was compared with UFH plus GPIIb/IIIa inhibitor in patients undergoing elective or urgent PCI in the Randomized Evaluation in PCI Linking Angiomax to Reduced Clinical Events 2 (REPLACE-2) trial.52 Although there were no differences in the overall primary composite endpoint of death, MI, urgent repeat revascularisation and in-hospital major bleeding at 30 days, analysis of the individual components revealed a significant reduction in the rates of in-hospital major and minor bleeding in the bivalirudin arm.
The use of bivalirudin was tested in 13,819 patients presenting with moderate-to high-risk NSTE-ACS planned for an invasive strategy in the Acute Catheterization and Urgent Intervention Triage Strategy (ACUITY) trial.53 Patients were randomised to receive one of three treatments: UFH or LMWH plus GPIIb/IIIa inhibitor, bivalirudin plus GPIIb/IIIa inhibitor or bivalirudin with bailout use of GPIIb/IIIa inhibitor. Those receiving bivalirudin were given a dose of 0.1 mg/kg IV bolus, followed by an infusion of 0.25 mg/kg/h. If patients underwent PCI, a further IV bolus of 0.5 mg/kg bivalirudin was given and the infusion dose was increased to 1.75 mg/kg/h prior to PCI and stopped at the end of the procedure. There was no significant difference in the rates of the composite endpoint of death, MI or unplanned revascularisation for ischaemia at 30 days between the two groups. The use of bivalirudin with GPIIb/IIIa inhibitor as a bailout strategy was also shown to be non-inferior compared with the combination of UFH/LMWH and a GPIIb/IIIa inhibitor. However, ischaemic events were significantly more common in bivalirudin-treated patients if they had not received pre-treatment with clopidogrel.54,55
A sub-study of the ACUITY trial assessed outcomes when patients were switched from UFH or LMWH to bivalirudin monotherapy at the time of PCI against those who received consistent UFH or LMWH.56 Death, MI and unplanned revascularisation rates were similar between the two groups, but there was significantly less major bleeding (2.8 % vs. 5.8 %, p<0.01) and an improvement in the net clinical benefit (defined as major adverse cardiovascular events plus bleeding) in patients who switched to bivalirudin. Qualitatively, similar observations were made in the Intracoronary Stenting and Anti-thrombotic Regimen– Rapid Early Action for Coronary Treatment (ISAR-REACT) 4 study. In patients presenting with NSTE-ACS undergoing PCI a significant reduction in bleeding was seen in patients treated with bivalirudin versus abciximab and UFH without a significant difference in death, recurrent MI or urgent target vessel revascularisation.57
A direct comparison of bivalirudin versus UFH in stable coronary artery disease was carried out in the ISAR-REACT 3 study.58 In 4,750 patients undergoing PCI for biomarker-negative NSTE-ACS, rates of death, MI and revascularisation at 30 days were similar between the two groups. A significant reduction in the rate of bleeding events was noted in the bivalirudin arm.
The ESC recommends bivalirudin as an alternative to UFH plus GPIIb/IIIa inhibitors in patients presenting with NSTE-ACS undergoing early invasive revascularisation, particularly if bleeding risks are high.4
Bivalirudin use following streptokinase has been demonstrated to significantly reduce rates of recurrent MI, but had no impact on mortality rates compared with UFH.59 An increase in bleeding rates was noted in the bivalirudin arm, but this was not significant.
In the Minimizing Adverse Hemorrhagic Events by Transradial Access Site and Systemic Implementation of Angiox (MATRIX) trial, 7,213 patients presenting with an ACS and planned for PCI were randomly assigned to receive either a bivalirudin infusion post PCI or UFH.60 Major adverse cardiac event and net adverse clinical events rates were not significantly different between the groups.
In the How Effective are Antithrombotic Therapies in Primary Percutaneous Coronary Intervention (HEAT-PPCI) trial, 1,812 patients presenting with STE-ACS either received bivalirudin of heparin following randomisation.61 Rates of major adverse ischaemic events in the setting of PPCI were significantly lower in the heparin group without an increase in bleeding rates.
In the Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction (HORIZONS-AMI) trial, 3,602 patients presenting within 12 hours of STE-ACS onset were randomised to receive either UFH plus GPIIb/IIIa inhibitor or bivalirudin.62 Rates of major bleeding, death and all-cause death were significantly reduced in patients receiving bivalirudin. An increase in rates of acute stent thrombosis was noted in the bivalirudin group, but this effect disappeared by 30 days.
ESC guidelines for STEMI recommend bivalirudin with bailout GPIIb/IIIa inhibitors over UFH plus GPIIb/IIIa inhibitors.23 An antithrombin agent should be given to patients presenting within 12 hours of symptom onset that have not been given reperfusion therapy.
New anticoagulant agents
Newer anticoagulants in the setting of ACS mostly target secondary prevention rather than the initial phase of the disease. These include anti-Xa therapies (apixaban, rivaroxaban, otamixaban) and the direct thrombin inhibitor dabigatran.
Phase III trials with anti-Xa drugs (apixaban and rivaroxaban) have shown a dose-related increase in the rate of bleeding when added to standard dual antiplatelet therapy. There was a trend towards a reduction in ischaemic events seen in patients treated with aspirin only. The Apixaban for Prevention of Acute Ischemic Events 2 (APPRAISE-2 trial) was stopped prematurely due to excessive bleeding with the apixaban regimen.63
Significantly lower rates of cardiovascular death were seen in patients with ACS established on aspirin and clopidogrel who were given low-dose rivaroxaban over placebo in the Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Standard Therapy in Subjects with Acute Coronary Syndrome–Thrombolysis in Myocardial Infarction 51 (ATLAS ACS 2-TIMI 51) study.64 This has led to the recommendation that the use of rivaroxaban 2.5 mg twice daily might be considered in combination with aspirin and clopidogrel if ticagrelor and prasugrel are not available for patients with NSTEMI who have high ischaemic and low bleeding risks.
Dabigatran was investigated in a Phase II dose-finding trial (Randomized Dabigatran Etexilate Dose Finding Study In Patients with Acute Coronary Syndromes Post Index Event With Additional Risk Factors For Cardiovascular Complications Also Receiving Aspirin And Clopidogrel [RE-DEEM]).65 Dabigatran, in addition to dual antiplatelet therapy, was associated with a dose-dependent increase in bleeding events and significantly reduced coagulation activity in patients with a recent MI.
A Phase III trial of the intravenous anti-Xa drug otamixaban did not reduce ischaemic event rates, but significantly increased bleeding rates when compared with UFH plus eptifibatide.66 These findings did not support the use of otamixaban for patients with NSTE-ACS undergoing planned early PCI.
Conclusion
The use of anticoagulant therapy is an essential adjunct to antiplatelet therapy in the acute treatment of ACS, and is limited to treatment during initial hospitalisation and revascularisation. Large, randomised clinical trials have shown the benefit of fondaparinux as a safer (with similar efficacy) alternative to either LMWH or UFH and it is the anticoagulant of choice on admission.
Once a decision is made for invasive management then either UFH or LMWH must be given during catheterisation to prevent formation of thrombus during the procedure. The role of bivalirudin in ACS has been controversial. It is an effective, but expensive drug with a short half-life; however, recent data showing an increase in acute stent thrombosis have largely negated the reduction seen in major bleeding rates.
A patient-centred approach is required to balance ischaemic and bleeding risk and it would appear that this can be successfully achieved with a choice of antiplatelet agents of differing potency and anticoagulants limited to fondaparinux and low dose heparins. At the time of writing, the newer direct anti-Xa and direct thrombin inhibitors lack the data to recommend routine use in ACS patients but may have a role in ACS patients presenting with persistent atrial fibrillation.