








Dr Kapur is an Associate Professor in the Department of Medicine at Tufts Medical Center. His research focuses on acute and chronic heart failure, circulatory support device development and cardioprotective mechanisms in the setting of acute myocardial infarction.
Dr Kapur began by considering the reasons for the paradigm shift in interventional therapy targeting ischaemia-reperfusion injuries. The past decade has seen a transition in outcomes for patients presenting with acute myocardial infarction (AMI). A cohort study (n=7,733) of older patients with first myocardial infarction (MI) showed that although the in-hospital mortality decreased by 28 % over 5 years, the 5-year incidence of heart failure increased by 25 %.1 The burden of ischaemic heart failure has become the new challenge in the treatment of MI. The current treatment paradigm focuses on rapid reperfusion to limit myocardial damage. However, the significant improvement in door-to-balloon times (DBTs) in the past 10 years (to <90 minutes) has had no impact on mortality rates, which remain at around 7 % for patients with anterior MI and 27 % for those with cardiogenic shock.2
Dr Kapur maintained that reperfusion has become a double-edged sword: while prolonged ischaemia can cause substantial injury, restoration of perfusion to the ischaemic heart can exacerbate tissue damage. DBTs have decreased because of the adage that ‘time is muscle’. When a coronary vessel becomes occluded, the acute ischaemic insult activates a process within cardiac myocytes involving a decrease in oxidative phosphorylation and adenosine triphosphate synthesis, which leads to a decrease in intracellular pH due to calcium influx and lactate elevations within these myocytes. This can lead to downstream effects on coronary function whereby the mitochondria become dysfunctional, generate reactive oxygen species and may even burst, resulting in cell death and necrosis. The current treatment of reperfusion creates a feed-forward mechanism, inducing further mitochondrial and oxidative damage. However, the body has an inherent counter-regulatory mechanism. First, at the endothelial level, endogenous tissue plasminogen activator attempts to autolyse thrombotic occlusions. At the cardiac myocyte level, there is a significant increase in salvage kinase activation, largely of extracellular-regulated kinase and protein kinase B. These kinases initiate an antiapoptotic signalling pathway designed to counteract the effect of reperfusion injury.
It is clear that many barriers exist to cardioprotective approaches and numerous drug trials attempting cardioprotection in the setting of AMI have failed. The critical barriers to success have been the mandate for rapid coronary reperfusion (i.e. DBT), the inability to target multiple cascades affecting reperfusion injury and the challenges of managing haemodynamic instability. A recent editorial critically appraised current approaches and asked the question: is it time to give up on cardioprotection?3 Pre-ischaemic conditioning is not feasible, post-ischaemic conditioning has limited feasibility, and studies have been inconclusive. Remote ischaemic conditioning has been associated with limited efficacy and findings have not been conclusive. There is therefore a need for new approaches to cardioprotection in AMI.
A key theme of the A-CURE meeting is the novel paradigm of limiting myocardial ischaemia by minimising oxygen demand then restoring oxygen supply, a procedure that has been termed functional reperfusion. Insights from the surgical management of ST-elevation MI have taught us that a procedure beginning with unloading (cardiopulmonary bypass) followed by reperfusing ischaemic myocardium results the in restoration of coronary function.4 However, this surgical mindset contrasts with the interventional approach and has not been adopted due to our limited ability to unload the myocardium without major cardiac surgery.
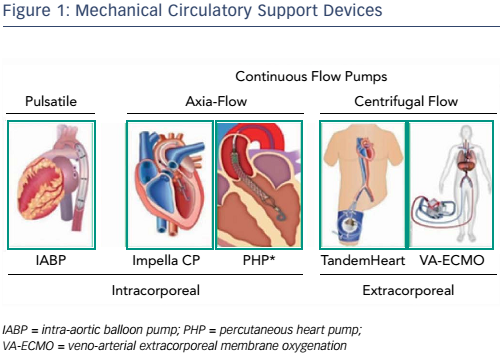
Over the past decade, a number of percutaneous mechanical circulatory support devices have become available (see Figure 1) and these have been described in previous presentations. The earliest work with intra-aortic balloon pumps showed that initiating mechanical support during infusion and reperfusion can reduce infarct size.5 This model is limited, however, in that the device is activated at the onset of ischaemia and remains on throughout reperfusion, so it is hard to translate to clinical use. Further studies have used axial-flow pumps (Impella®, Abiomed) as a direct left ventricular (LV) loading mechanism and focussed not only on activation timing but also investigated the concept of total and partial unloading. An early study in sheep found that if Impella is activated during reperfusion alone, the degree of unloading correlates with a reduction in infarct size.6
Dr Kapur’s team has been investigating the novel hypothesis that initially reducing LV work and delaying coronary reperfusion may limit myocardial injury in AMI. The choice of delayed reperfusion was driven by necessity in order to replicate events in the catheterisation laboratory. The study employed the TandemHeart® (CardiacAssist Inc) device, which requires transseptal implantation and the use of two large cannulas that take time to implant. Fortuitously, the delayed reperfusion proved to be one of the critical components in translating acute unloading to the setting of AMI. The study used a closed chest swine model, again replicating typical events in the catheterisation laboratory. In the MI group (n=4), MI was induced by occlusion of the left anterior descending (LAD) artery for 120 minutes, followed by 120 minutes of reperfusion without mechanical support. In the mechanically-supported group (MI plus unload; n=4), percutaneous left atrial-to-femoral artery bypass was initiated after 120 minutes of ischaemia, and LAD artery occlusion was prolonged for an additional 30 minutes, followed by 120 minutes of reperfusion with device support. A significant reduction in infarct size was seen, which correlated with the reduction of LV stroke work.7
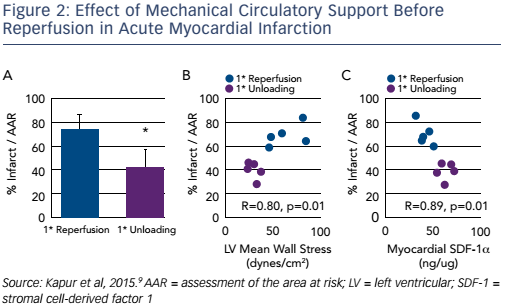
An editorial, published alongside this article, posed a number of questions. First, is it clinically feasible to use devices requiring transseptal implantation in the setting of AMI? What is the optimal timing of delayed reperfusion? What was the mechanism responsible for the beneficial effects?8 These questions formed the basis of Dr Kapur’s next studies. At the time he had been studying the feasibility of left atrial unloading compared with LV unloading and concluded that the Impella provided the most effective unloading signature, giving a reduction in LV pressure and volume. In addition, the Impella device eliminated the need for transseptal puncture arterial access. A study was designed to test the hypothesis that initially reducing LV work and extending the delay to coronary reperfusion may limit myocardial injury in AMI. In the primary reperfusion group, the LAD artery was reperfused for 120 minutes. In the primary unloading group, after 90 minutes of ischaemia the axial flow pump was activated and the LAD artery left-occluded for an additional 60 minutes, followed by 120 minutes of reperfusion. There was a significant 43 % reduction in infarct size in the primary unloading group compared to primary reperfusion (73 ± 13 % versus 42 ± 8 %; p=0.005). There was a correlation between infarct size reduction and LV wall stress.
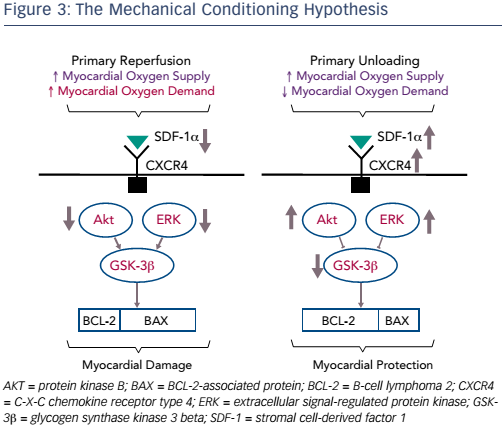
Another interesting finding was that unloading activates biological cardioprotective processes, increasing myocardial levels of the chemokine stromal cell-derived factor (SDF)-1 alpha, and that the regression plot of infarct size against SDF-1 alpha levels in the myocardium was almost linear (R=0.89; p<0.01; see Figure 2).9 This study led to the mechanical conditioning hypothesis: first unloading the LV, then delaying reperfusion activates a cardioprotective programme that limits myocardial damage in AMI (see Figure 3).9
This hypothesis prompted further questions: how important is the delayed reperfusion? How important is the role of the kinases (i.e. is the mechanism primarily haemodynamic or biological)? What is the long-term effect of this intervention on LV recovery? Dr Kapur’s team is addressing these questions in a number of ongoing studies. In one currently unpublished study, a series of animals were divided into groups with ischaemia reperfusion alone or a delay in reperfusion after activating the Impella device of 15, 30 and 60 minutes. In the final group, the Impella device was activated after balloon reperfusion. Results showed that 15–30 minutes of primary unloading was required to achieve infarct size reduction. A further study investigated the biological mechanisms for this finding and found that delayed reperfusion is required to activate cardioprotective signalling involving SDF-1 alpha. In another set of studies, following occlusion of the LAD artery, C-X-C chemokine receptor type 4 inhibitors (which inhibit SDF-1 alpha influx) or kinase inhibitors were administered. Both interventions increased the infarct size, demonstrating that loss of kinase function attenuates the cardioprotective effect of primary unloading. In the final study, SDF-1 alpha was administered via intracoronary delivery to try to augment its cardioprotective effect. However, it was not possible to further reduce the infarct size by this method.
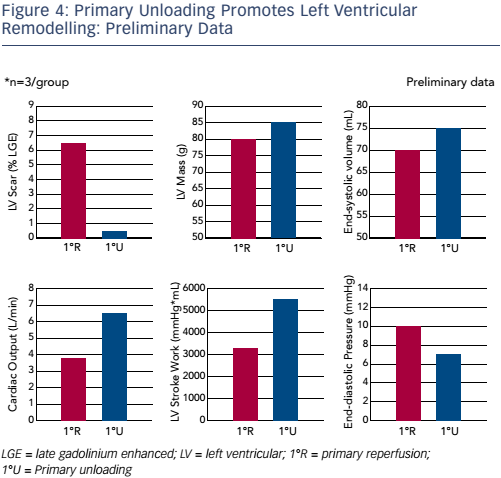
The final question – does acute unloading impact LV recovery? – was answered in a study with a 28-day follow-up period. In the primary reperfusion group, the infarct scar at 28 days was significantly larger than that in the primary unloading group. In terms of function, the primary reperfusion group showed a characteristic LAD artery infarct pattern whereas the primary unloading group showed primarily intact myocardium. Preliminary data (n=3) showed significant reductions in LV scar (see Figure 4).
In summary, there is a point at which the injury clock can be stopped and a protective process initiated that will limit the damage once reperfusion begins. This is the focus of Dr Kapur’s future work.
Young Investigator Scholarship Presentation
Michele Esposito, a member of Dr Kapur’s research team, presented the abstract that won the Young Investigator Scholarship awarded by the A-CURE Working Group. This study tested the hypothesis that primary unloading promotes myocardial salvage in AMI through regulation of gene expression within the infarct zone. The LAD artery of male pigs (n=4/group) was occluded for 90 minutes. In the primary reperfusion group, the LAD artery was reperfused for 120 minutes. In the primary unloading group, after 90 minutes of ischaemia a mechanical circulatory support device was activated and the LAD artery left-occluded for an additional 30 minutes, followed by 120 minutes of reperfusion. Myocardial infarct size was quantified by triphenyl tetrazolium chloride staining. Whole-transcript expression analysis was performed using a porcine microarray platform.
Quantitative polymerase chain reaction confirmed the expression of select genes from regulated pathways. Scanning electron microscopy evaluated mitochondrial integrity within infarct zones. Sham operated LV samples were used as controls.
Consistent with previous studies, primary unloading reduced infarct size compared to reperfusion alone, from 65 % to 34 %. Gene expression analysis yielded a heat map representing the 2,200 genes significantly regulated by primary reperfusion or primary unloading (p<0.01). A significant shift in gene expression was seen: the primary unloading group showed a heat map similar to the sham controls. The investigators then identified a number of key regulatory pathways altered by primary unloading, including inflammatory and fibrotic pathways. Specifically, matrix metallopeptidases MMP2 and MMP9 were upregulated in the reperfusion group and downregulated in the unloading group. MMP2 and MMP9 are involved in the breakdown of extracellular matrix, leading to adverse remodelling, and are markers of inflammatory response.
In the reperfusion group, there was increased expression in SMAD3, an intracellular signal transducer and transcriptional modulator that, following mechanical stretch, is phosphorylated by transforming growth factor-beta, and then converts fibroblasts to myofibroblasts, increasing adverse remodelling. Expression of genes in the electron transfer chain, which is responsible for adenosine triphosphate synthesis and other important pathways involved in cellular metabolism, was decreased in the reperfusion group compared with the unloading and sham groups, suggesting that unloading preserves the integrity of the electron transfer chain during AMI.
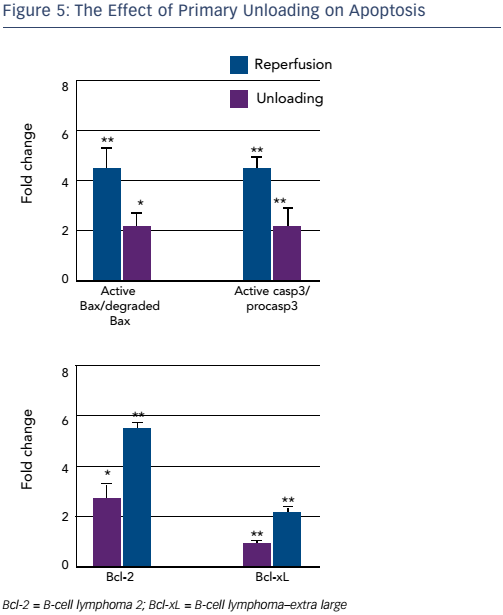
Finally, examination of mitochondrial integrity revealed a significantly increased number of intact mitochondria per cardiac myocyte in the unloading versus reperfusion group. Since mitochondrial function is linked to apoptosis, key components of the apoptotic pathway were examined. A higher density of the pro-apoptotic active BAX protein and procaspase-3 were seen in the reperfusion group, whereas higher densities of degraded, inactive, BAX and caspase-3 were seen in the sham and unloading groups. Higher levels of antiapoptotic agents B-cell lymphoma 2 and B-cell lymphoma–extra large were identified in the unloading group (see Figure 5).
In summary, this study identified for the first time that primary unloading triggers a global shift in gene expression within the infarct zone that is associated with preserved mitochondrial integrity and cellular respiration, reduced apoptosis, inflammation and fibrosis during the acute phase of MI. These data suggest that unloading the left ventricle and delaying reperfusion promotes cardioprotective signalling and may be a novel approach to limiting myocardial damage during AMI and preventing the subsequent development of ischaemic heart failure.
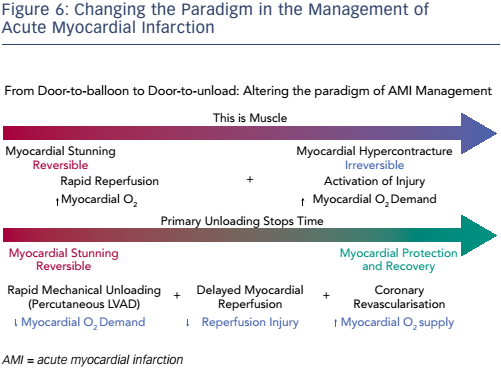
Dr Kapur concluded by stating that the burden of ischaemic heart failure will grow and that new approaches to cardioprotection in AMI are needed. Primary unloading reduces infarct size through a two-pronged approach: first by reducing the wavefront of MI and second by activating a cardioprotective process (see Figure 6). However, prospective randomised controlled studies are required to test the clinical validity of these preclinical findings.